Adaptive bioinformatic solutions
Empowering biotechnology and pharmaceutical innovation through bespoke software, intelligent data analysis, and precision assay design.
2012
80+

About us
Alternate Allele Consulting was founded over a decade ago when biotech veteran James ("J") Ireland realized that neither internal teams nor traditional consulting firms could deliver the responsive, high-depth bioinformatics support that modern R&D requires. To fill that gap, J created a consulting practice built for the realities of today's biotechnology landscape—fast-moving, data-intensive, and constantly evolving.


We focus on delivering
Right-sized solutions
We tailor our services to perfectly fit every client, serving all organizations from large pharmaceutical firms to lean, founder-led startups.
Cross-disciplinary expertise
Our team unifies expertise spanning bioinformatics, statistics, software engineering, and essential laboratory workflows.
Adaptive, forward design
Our designs are built to evolve, readily adapting to incorporate new technologies, data types, and emerging scientific needs.
Our clients
Our clients span the full spectrum of modern R&D — more than 80 companies across biotech, pharma, tech, and academia. We've partnered with half of the world's top ten pharmaceutical companies, as well as publicly traded biotechs and pre-seed startups. Along the way, we've served as external experts, embedded team members, co-founders, stopgaps, and, when needed, a computational rescue team.











How can we help you?
We offer three core services designed to accelerate your research and development.
Tailored Digital Infrastructure
Custom bioinformatics software tailored to your workflow. From pipeline automation to interactive data visualization tools, we build solutions that integrate seamlessly with your existing infrastructure.
- Custom pipeline development
- Tool integration & automation
- Web-based analysis platforms
- Database design & management
Actionable Data Insights
Transform complex biological data into actionable insights. Our expertise spans statistical analysis, machine learning, and AI-driven approaches to unlock patterns in your data.
- Genomic & transcriptomic analysis
- Machine learning & AI modeling
- Statistical consulting
- Predictive analytics
Robust Assay Engineering
Precision-engineered assays for your specific research needs. We combine computational design with experimental knowledge to create robust, reproducible assays.
- Primer & probe design
- Assays for diverse organisms (human, model species, microbes, viruses)
- Applications including genotyping, CNV detection, enrichment, pathogen detection
- Multiplexing strategies
News
Publications, preprints, and collaborative research from our team.
New Protocol Contribution in SpringerMethods: RNA Gene Expressions for Stroke Etiology
New Protocol Contribution in SpringerMethods: RNA Gene Expressions for Stroke Etiology
We are pleased to announce our contribution to a newly published methods chapter in the Springer Methods in Molecular Biology series. The chapter, titled "RNA Gene Expression to Identify the Etiology of Acute Ischemic Stroke: The Biomarkers of Acute Stroke Etiology (BASE) Study)", presents a validated workflow for using whole-blood gene expression to classify ischemic stroke etiology — a major clinical challenge where a substantial fraction of patients leave the hospital without a definitive cause.
The chapter outlines standardized protocols for sample handling, RNA expression profiling, and downstream computational analysis, providing a reproducible framework for translational stroke biomarker research. Our team supported analytical components of this work, contributing to the development of robust expression-based classifiers for stroke subtype.
Gene Expression Signature Developed by Alternate Allele Enables New Stroke Etiology Test
Gene Expression Signature Developed by Alternate Allele Enables New Stroke Etiology Test
Alternate Allele Consulting is proud to highlight our contribution to Ischemia Care's newly launched ISCDx test, a blood-based laboratory-developed test (LDT) designed to determine the underlying cause of acute ischemic stroke. This test, profiled today by GenomeWeb and 360Dx, represents an important step toward faster, more accurate stroke management—particularly for identifying cardioembolic versus large-artery etiologies, a distinction that can guide secondary prevention and reduce recurrent stroke risk.
AAC led the development and validation of the gene expression signature at the core of the ISCDx assay, mining data from the large, multi-center BASE Study (Biomarkers of Acute Stroke Etiology). BASE includes over 1,700 patients enrolled across more than 20 U.S. stroke centers and has demonstrated that genomic profiling of peripheral blood can help discriminate stroke mechanisms more rapidly than imaging alone, which still leaves nearly one-third of strokes classified as "unknown etiology".
The ISCDx test is now being deployed through a CLIA-certified laboratory and is undergoing continued clinical validation for differentiating acute ischemic stroke from transient ischemic attack and other stroke subtypes. By enabling rapid, mechanism-informed decision-making, this signature-based approach has the potential to improve treatment selection, reduce misclassification, and enhance secondary prevention strategies.
New Publication on Molecular Subtyping of Archival Bladder Cancers
New Publication on Molecular Subtyping of Archival Bladder Cancers
Alternate Allele Consulting is pleased to highlight our contribution to a collaborative study published today in PLOS ONE. This work presents a custom microfluidics-based gene expression panel designed to classify bladder tumors into clinically meaningful basal and luminal molecular subtypes—even when working with challenging archival FFPE samples.
In this study, researchers demonstrated that the panel reliably stratifies tumors across multiple public datasets and applied it to a new cohort of 204 bladder cancer samples, including non-muscle invasive disease, muscle-invasive tumors, and metastases. The analysis revealed clear subtype patterns: NMIBCs were largely luminal, metastases predominantly basal, and MIBCs split between the two groups. Mutational and FGFR3 protein-level data further reinforced known subtype biology.
Genome Coordinate Conventions
Genome Coordinate Conventions
Denoting a contiguous region in a reference genome seems straightforward enough. However, I've seen many a bioinformatician get tripped up by the different conventions and formats used on web sites, file formats and genomic tools. Here are some things to keep in mind.
Chromosome
Taking the human genome as our example, chromosome names may or may not use a prefix so chromosome 12 may be "chr12", "ch12" or simply "12". Also, the mitochondrial chromosome may be "MT" or simply "M" and the sex chromosomes might be denoted as "X" and "Y" or "23" and "24".
Strand
Typical conventions for denoting the plus vs minus strand orientation of a sequence include using "1" and "-1" or "+" and "-". It is worth noting that for most of the genomic resources I use, genomic positions are almost always given relative to the plus strand even if the feature is said to be on the minus strand. For example, BLAT-ing a minus strand sequence against the human genome at UCSC will return a search result with "-" strand but the start and stop positions will be given relative to the plus strand. Minus strand positions would start counting from 0 at the opposite end of the chromosome (q-arm).
Position
Several conventions exist which differ in (1) what number they start with when numbering bases, (2) whether they number the bases themselves or the spaces between bases and (3) whether the interval is considered "open" or "closed". The notion of "closed" and "open" intervals is a mathematical concept which in this context implies either that the start and stop of the interval should be included (a closed interval) or they should not be included (an open interval). Sometimes you will see square brackets used to denote closed intervals (e.g. [12,20]) and parentheses used to denote open intervals (e.g. (12,20)).
Below are examples of three of the more common conventions. In each case I show the coordinates of an ATG subsequence (in red) and a cut site (marked by a red triangle).
Base-counted, one-start (a.k.a. one-based, fully-closed)
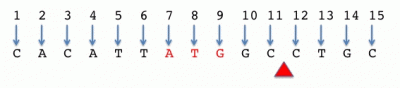
ATG location: 7-9 or [7,9]
Cut site: 11^12 or (11,12)
Interval length = stop - start + 1
Notes:
- This is by far the most common convention used by the major genome browsers, tools like BLAST and BLAT, etc.
- Using a base-counted system is problematic for describing features that occur between bases such as insertions or enzyme cut sites. To deal with this, some conventions replace the "-" usually used to separate the start and stop position with an alternative notation such as "^". Alternatively, one could use the parentheses notation.
- Base-counted systems can short-hand a one base interval with just the start (or stop) location. This is useful for denoting the location of SNPs, for example.
Space-counted, zero-start
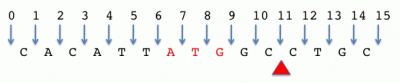
ATG location: 6-9
Cut site: 11-11
Interval length = stop - start
Notes:
- Less common, this convention is attractive because of its natural way of denoting features between bases such as insertions, etc. Length calculations are also simple.
Zero-based, half-open
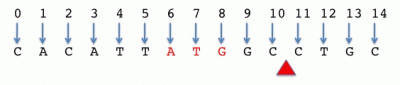
ATG location: 6-9 or [6,9)
Cut site: 11-11 or [11,11)
Interval length = stop - start
Notes:
- In this convention, the start base is included in the interval but not the stop base. This convention is used in data formats (especially at UCSC) such as BED.
- Although conceptually different, space-counted, zero-start and zero-based, half-open give the same start and stop coordinates for intervals.
- Python programmers will find this convention familiar as Python indexes arrays in the same manner.
Conclusions
As you can see, the differences between conventions are subtle, which makes it very easy to make undetected errors. In my experience, programmers prefer to use one of the zero-start conventions since most computer languages use zero-based indexing where as biologists are more fond of the one-start conventions. This commonly leads to a juggling of conventions where software using a zero-start convention is switched to one-start for displaying results to the user. The UCSC Genome Browser is a good example of a site with straddling conventions between the user interface and the underlying data tables and it must cause some confusion because they've devoted a FAQ entry to the issue.
My personal favorite convention is the space-counted, zero-start convention. All intervals require a start and stop location (even for single base features like SNPs), which makes up for in consistency what it might lack in conciseness. Denoting a location between two bases, such as for an insertion or enzyme cut site, is conceptually clear and does not require syntatic trickery like the base-counted methods. My understanding is that one of the many differences between the public and Celera human genome sequencing efforts was the public effort used base-counted, one-start while Celera used space-counted, zero-start although I don't have a reference to confirm this.
I haven't touched on the coordinate conventions used in the human variant world which give positions relative to cDNA. The HGVS variant nomenclature is a good example of this. Among the interesting features of this convention are starting counting (at 1) with the A of the ATG initiation codon, using negative coordinates and skipping the 0 base altogether. Good times.
References
- A great blog post from the Bergman lab dealing with genome coordinate conventions with an emphasis on transposable elements annotation.
- UCSC's description of the zero-based, half-open convention.
Welcome to Alternate Allele
Welcome to Alternate Allele
It seemed fitting to spend some time this Thanksgiving holiday to finally write a post about the launch of Alternate Allele Consulting. We officially opened doors on September 1, 2012 and haven't slowed down since. Like most small businesses, we've relied on friends, family and our extended networks to get things off the ground. I wanted to take a moment to recognize those folks now.
Alternate Allele's guiding principle is to do useful work and to do it thoughtfully and honestly. This blog reflects that mentality. We won't post unless we have something useful to say, which means we might not post that often. We hope that hearing about the decisions and people who helped us get started might help those considering a similar endeavor.
I'd like to thank Joel Westbrook of Miles and Westbrook for help with legal counsel and Jim Crofoot of Crofoot Accountancy Corp for accounting services. Hasto Anggoro designed our logo and business cards via 99designs. I'm especially proud of the IUPAC table we put on the back of our cards. Curtis Kautzer did a wonderful job taking photographs of our labware.

I would also like to thank Katie Kong of Kong and Associates and Andrew Boudreau of Gentelligent for providing business advice. Finally, I'm working with 5AM Solutions on some exciting projects and I'm grateful for their ongoing collaboration.
To get the business off the ground we've used Google Apps for email/calendar/docs, FreshBooks for invoicing and time tracking, BitBucket for source code repository management, SquareSpace for the website and 99designs for the logo.
Lets collaborate
Ready to solve your bioinformatics challenges? We'd love to hear from you.